
Impact of Surface Area on Sensitivity in Autonomously Reporting Sensing Hydrogel Nanomaterials for the Detection of Bacterial Enzymes

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
2.1. Materials
2.2. Methods
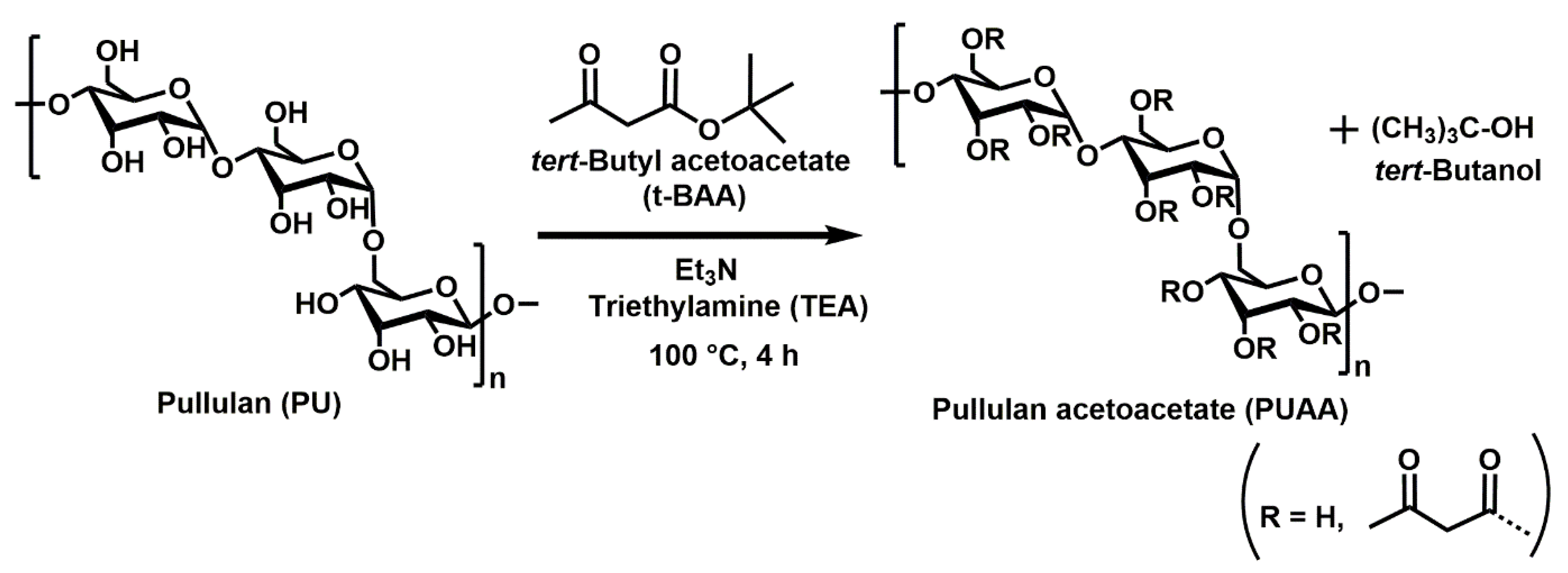
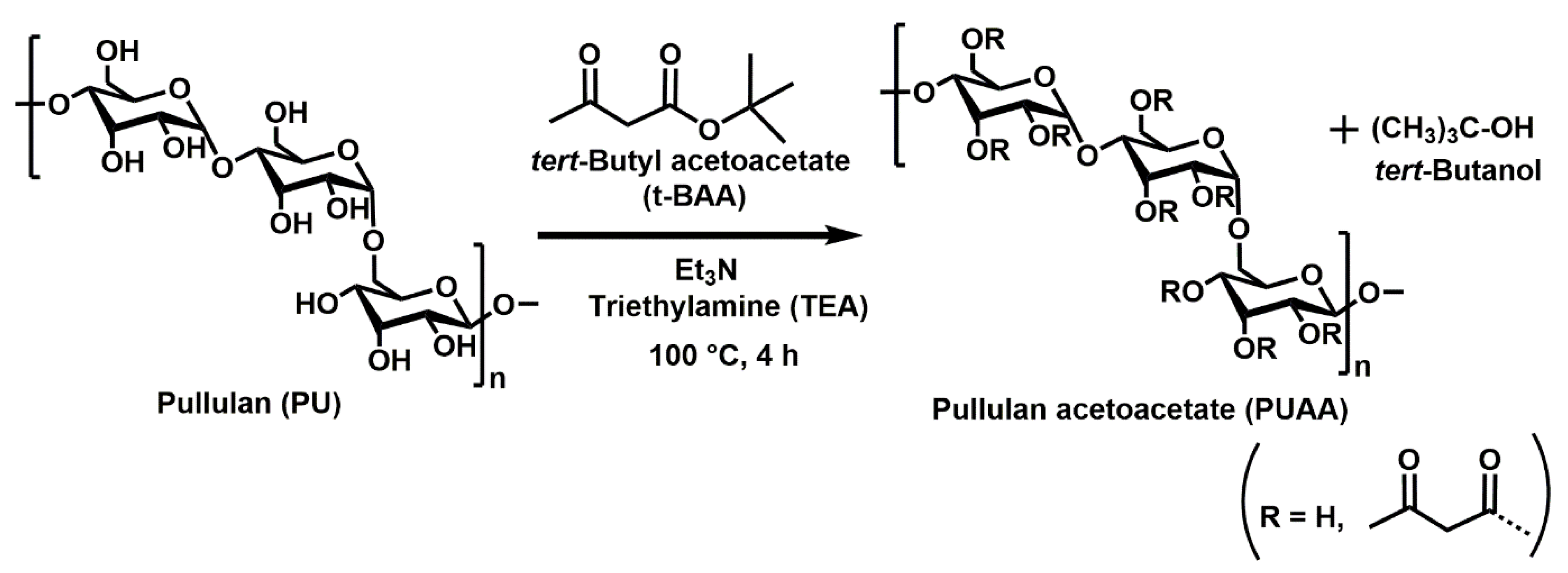
2.2.1. Preparation of Micro-Cuboid Structured (MS) Hydrogel Films
Preparation of Polydimethylsiloxane (PDMS) Mold
Synthesis of Gel Precursor and Fabrication of Hydrogel
Fabrication of Micro-Cuboid Patterned Hydrogel Film
2.2.2. Preparation of Chitosan/Polyethylene Oxide (CS/PEO) Nanofibers (NFs) and CS/PEO Hydrogels (HG)
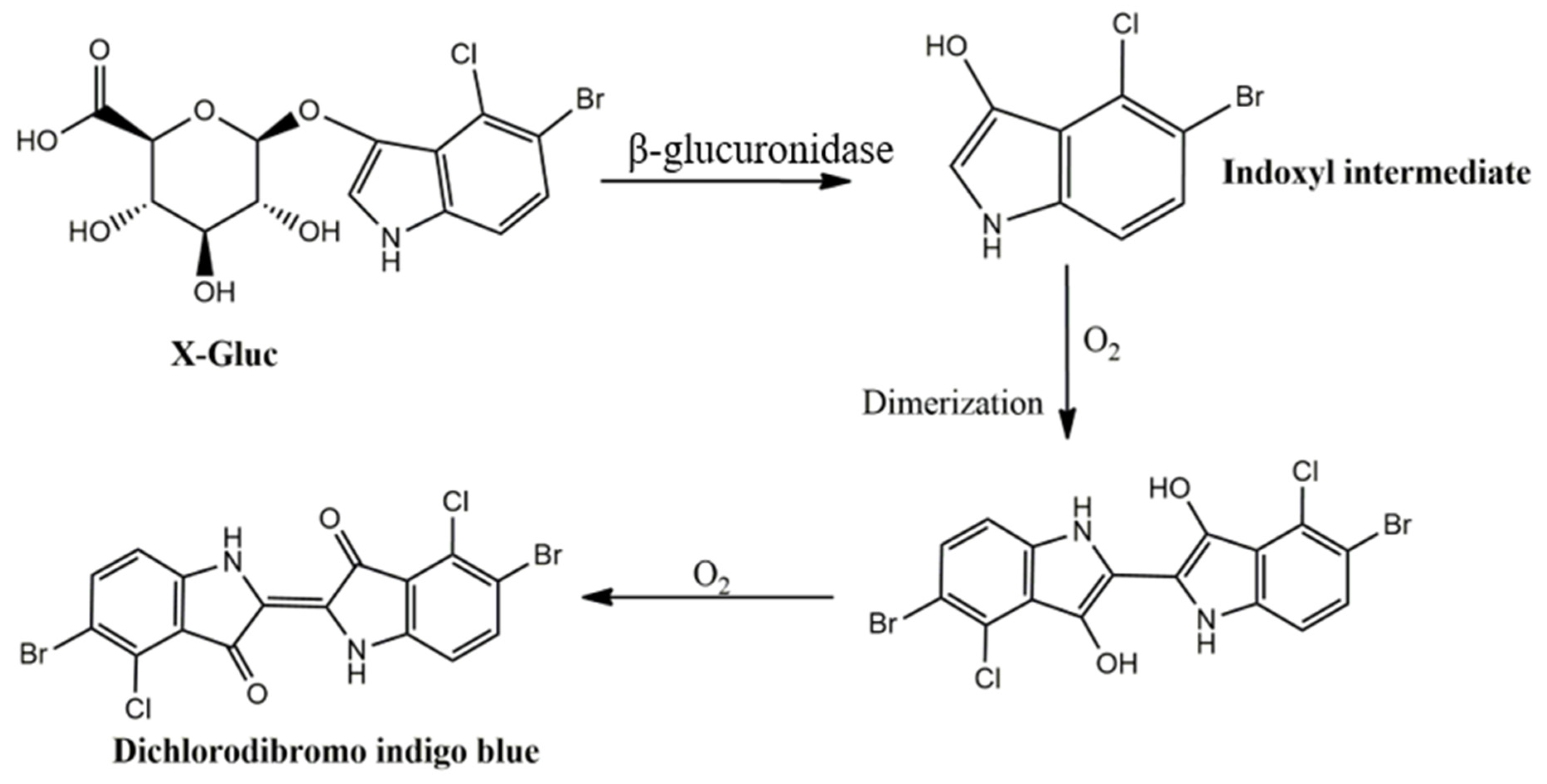
2.2.3. Surface Modification of Micropatterned Hydrogel Films and Nanofibers with the Chromogenic Substrate X-Gluc
Surface-Modification of PUAA-ADH Micro-Patterned Hydrogel Film with Chromogenic Substrate
Modification of CS Nanofibers and Hydrogels with Enzyme Substrate
2.2.4. Enzymatic Hydrolysis Study of Dye Labeled PUAA-ADH Hydrogels and CS/PEO Nanofibers
2.2.5. Enzymatic Hydrolysis in Bacterial Suspension Cultures
2.2.6. Determination of Limit of Detection (LOD) and Limit of Quantification (LOQ) for β-Glucuronidase in CS/PEO NFs and PUAA-ADH MS Hydrogel Films
2.3. Characterization of PUAA-ADH Hydrogel Films and CS/PEO Nanofibers
2.3.1. Scanning Electron Microscopy (SEM)
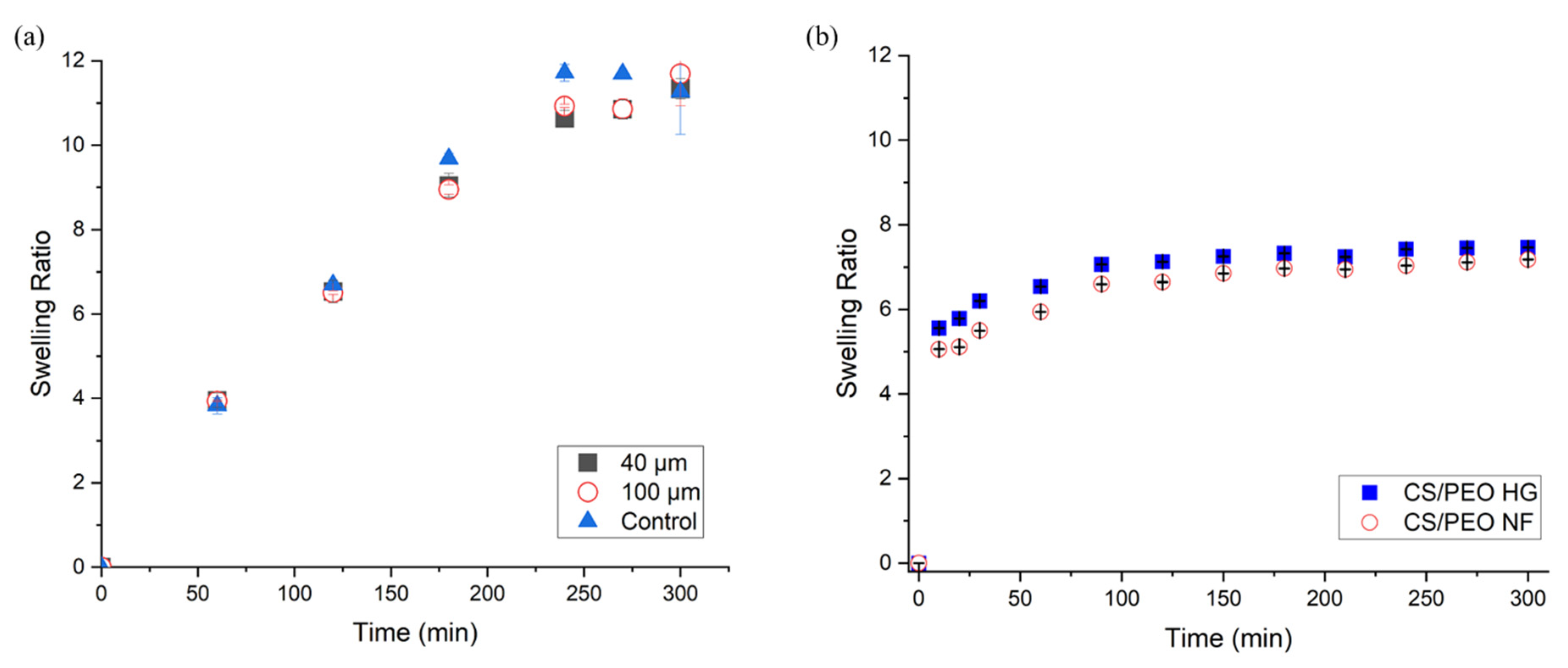
2.3.2. Determination of the Swelling Ratio of Dried Hydrogel Film
2.3.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
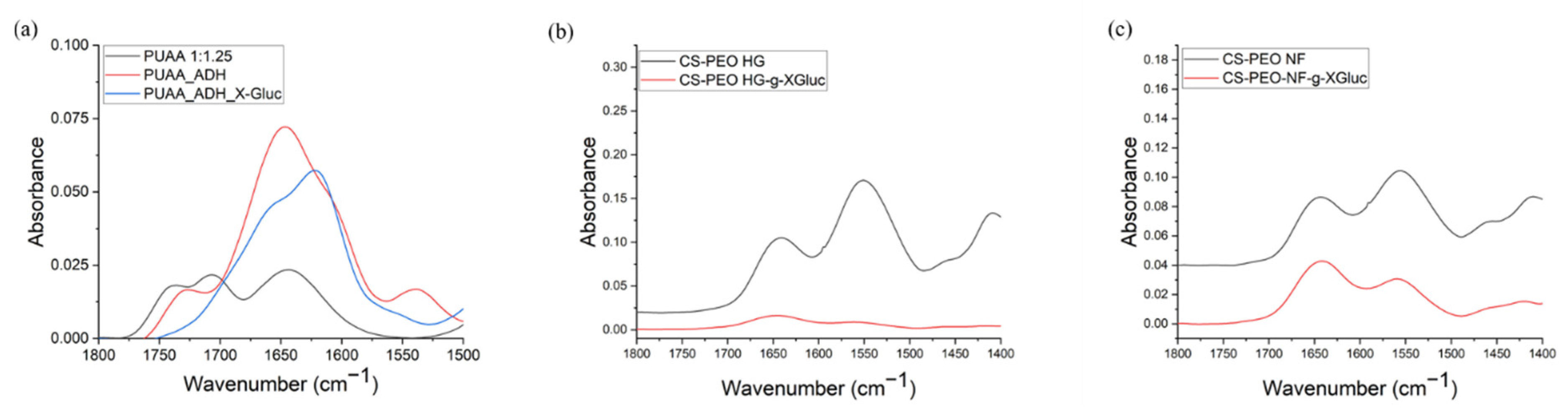
2.3.4. Fourier Transform Infrared (FTIR) Spectroscopy
2.3.5. UV-Vis Spectroscopy
3. Results and Discussion
3.1. Fabrication and Characterization of PUAA-ADH MS and CS/PEO NFs
3.1.1. Scanning Electron Microscopy
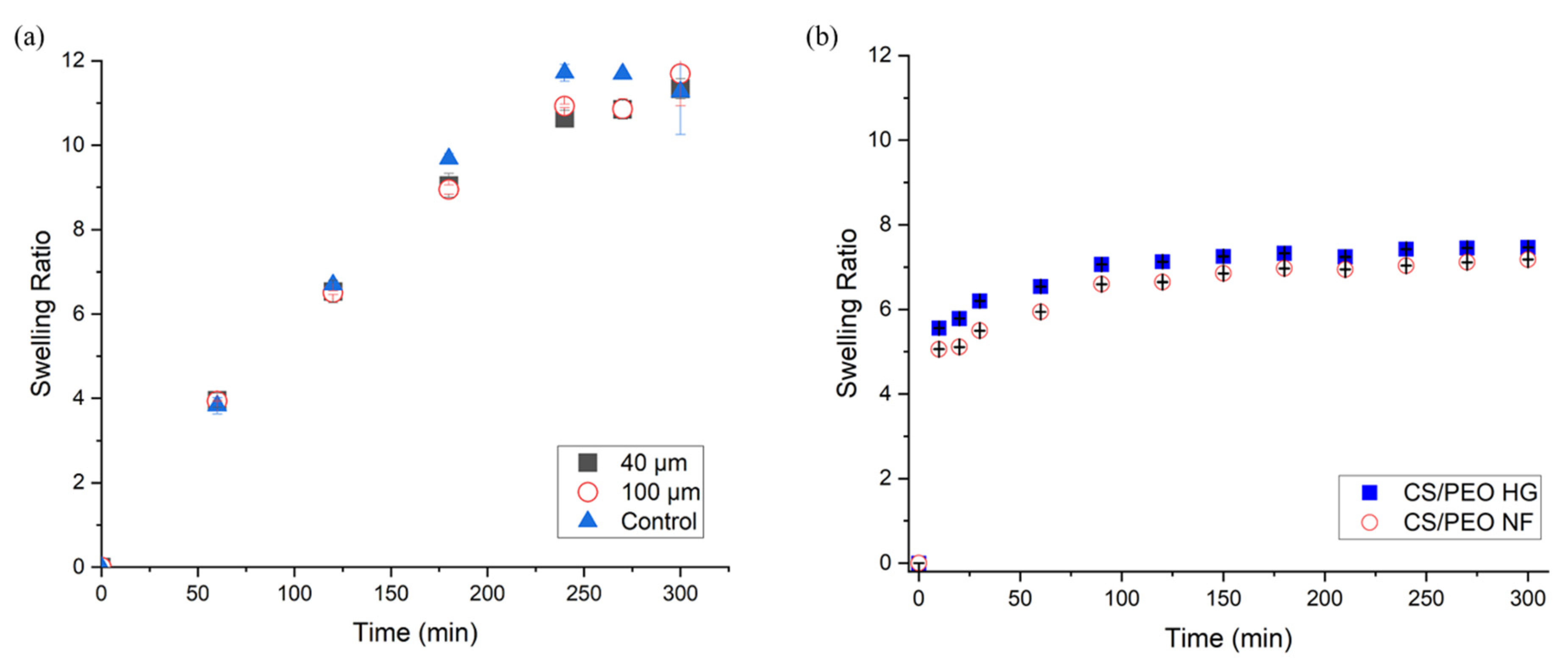
3.1.2. Swelling Ratio
3.1.3. FTIR Spectroscopic Analysis of Modified PUAA-ADH MS Films and CS/PEO NFs
3.1.4. Quantification of Pullulan Acetoacetate Functional Groups Using NMR Analysis
3.2. Enzymatic Reaction in ß-Glucuronidase Sensing PUAA-ADH Hydrogels
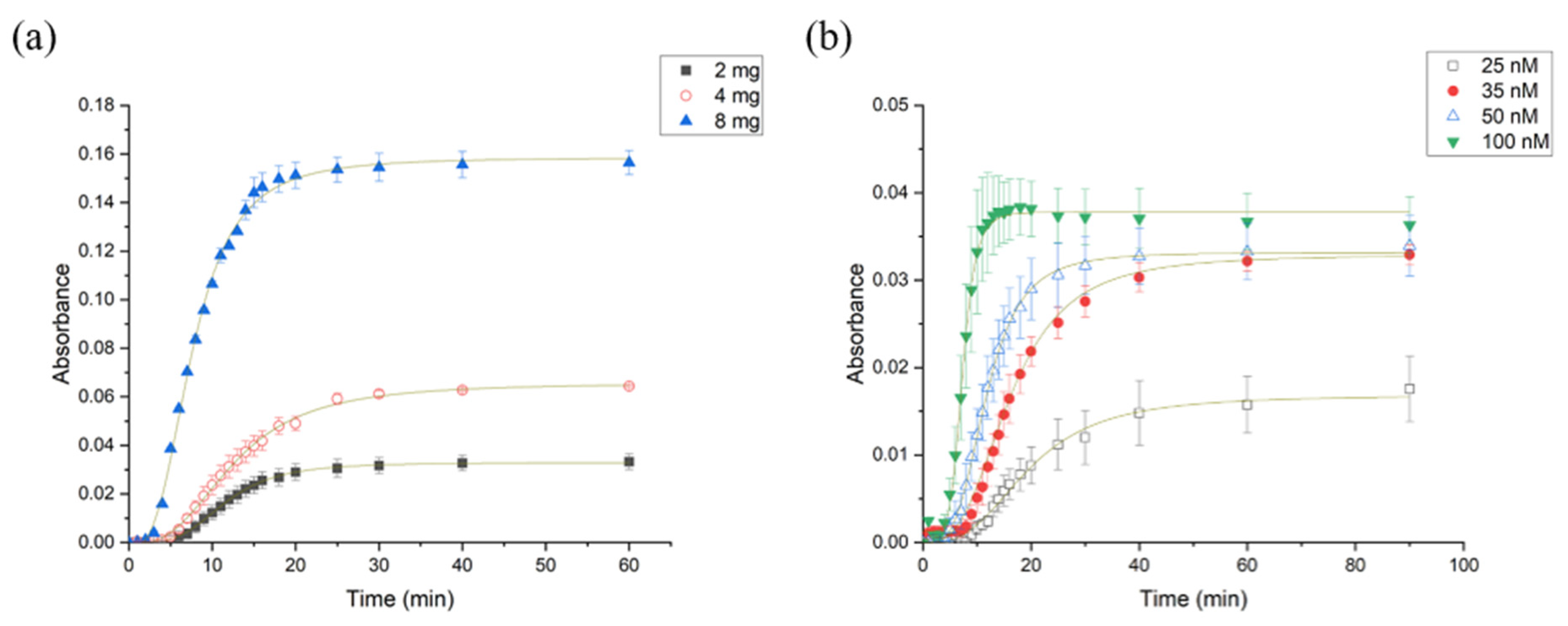
3.2.1. Impact of Varying Substrate and Enzyme Concentration on the Rate of Enzymatic Hydrolysis of X-Gluc Grafted PUAA-ADH
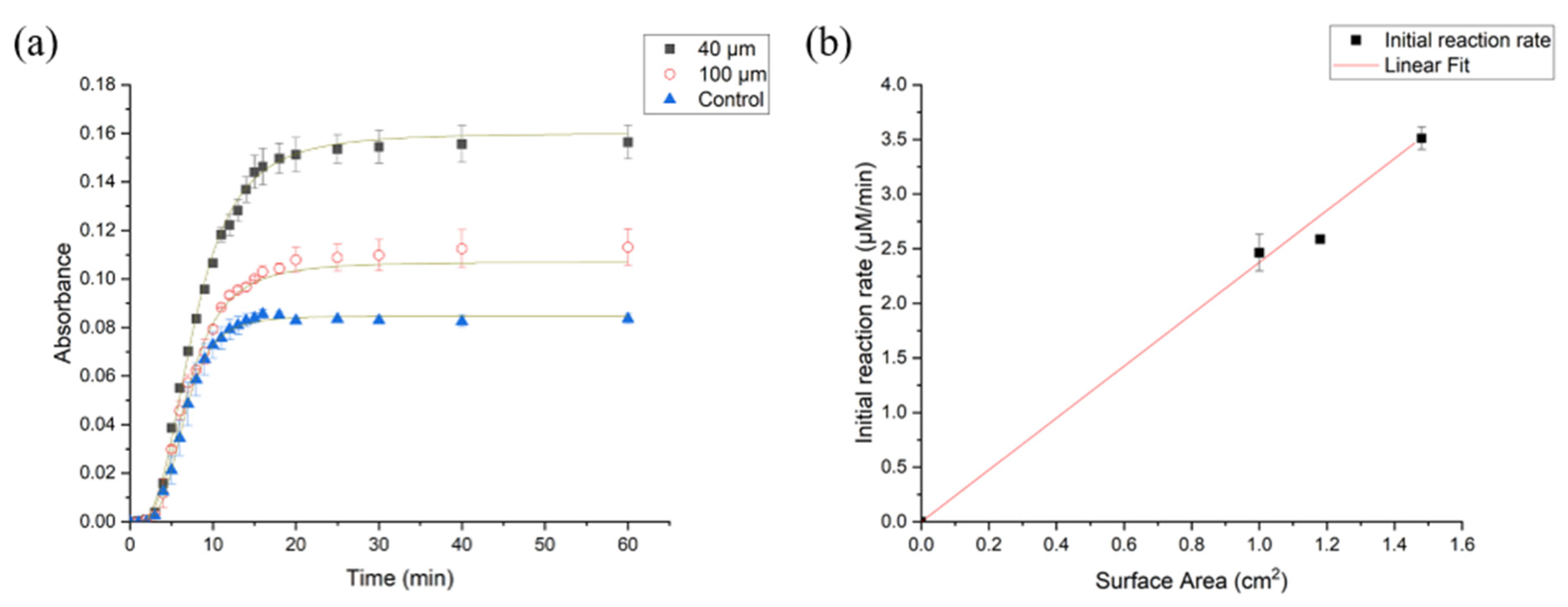
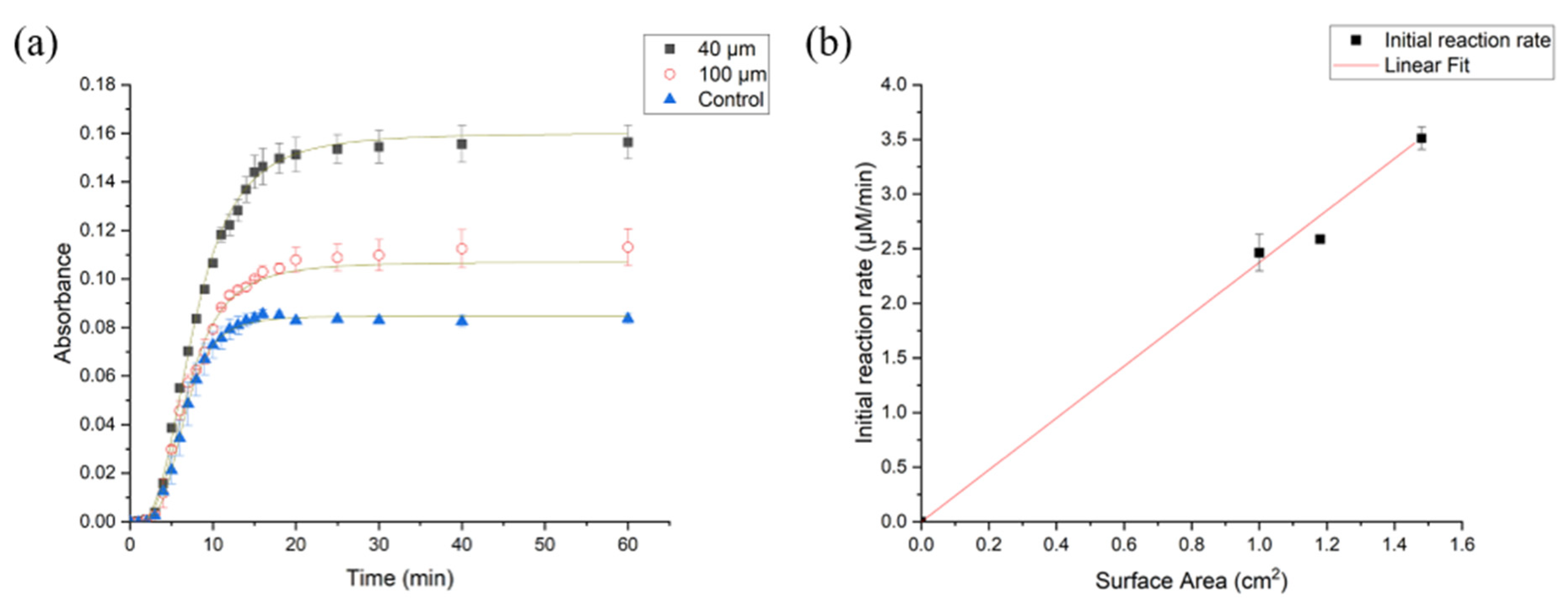
3.2.2. Impact of Varying Surface Area on the Rate of Enzyme Hydrolysis
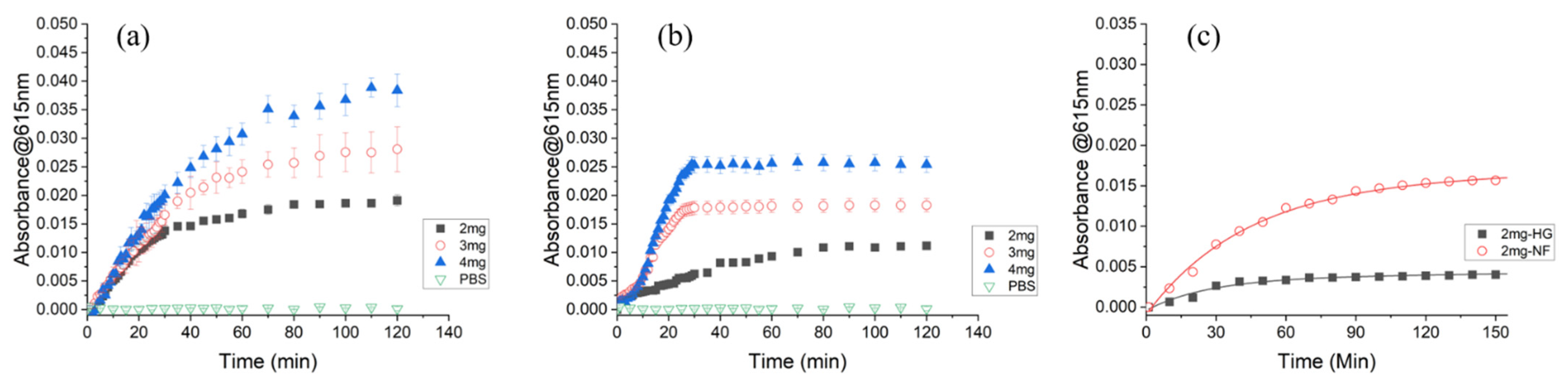
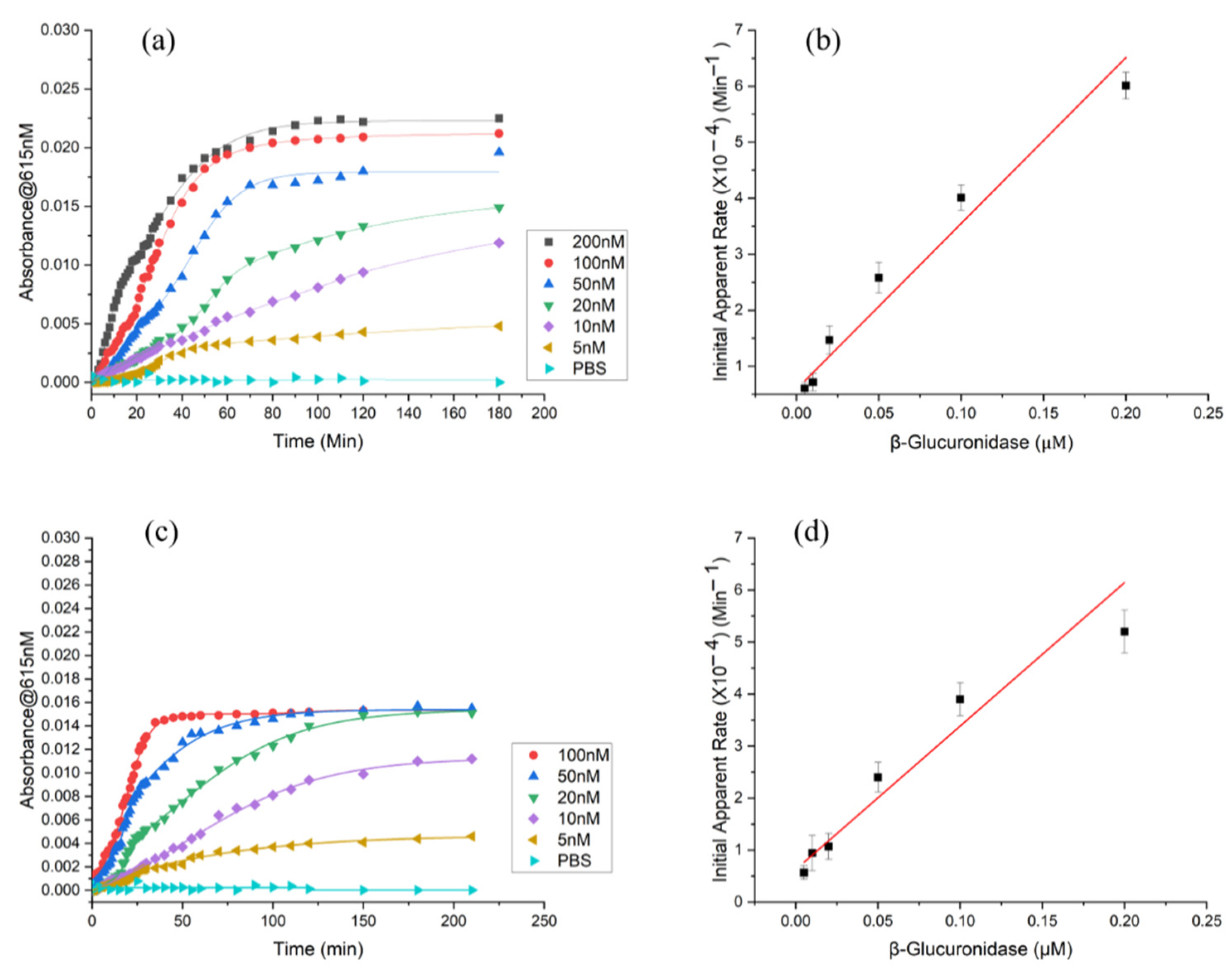
3.3. Enzymatic Reaction in ß-Glucuronidase Sensing CS/PEO NFs
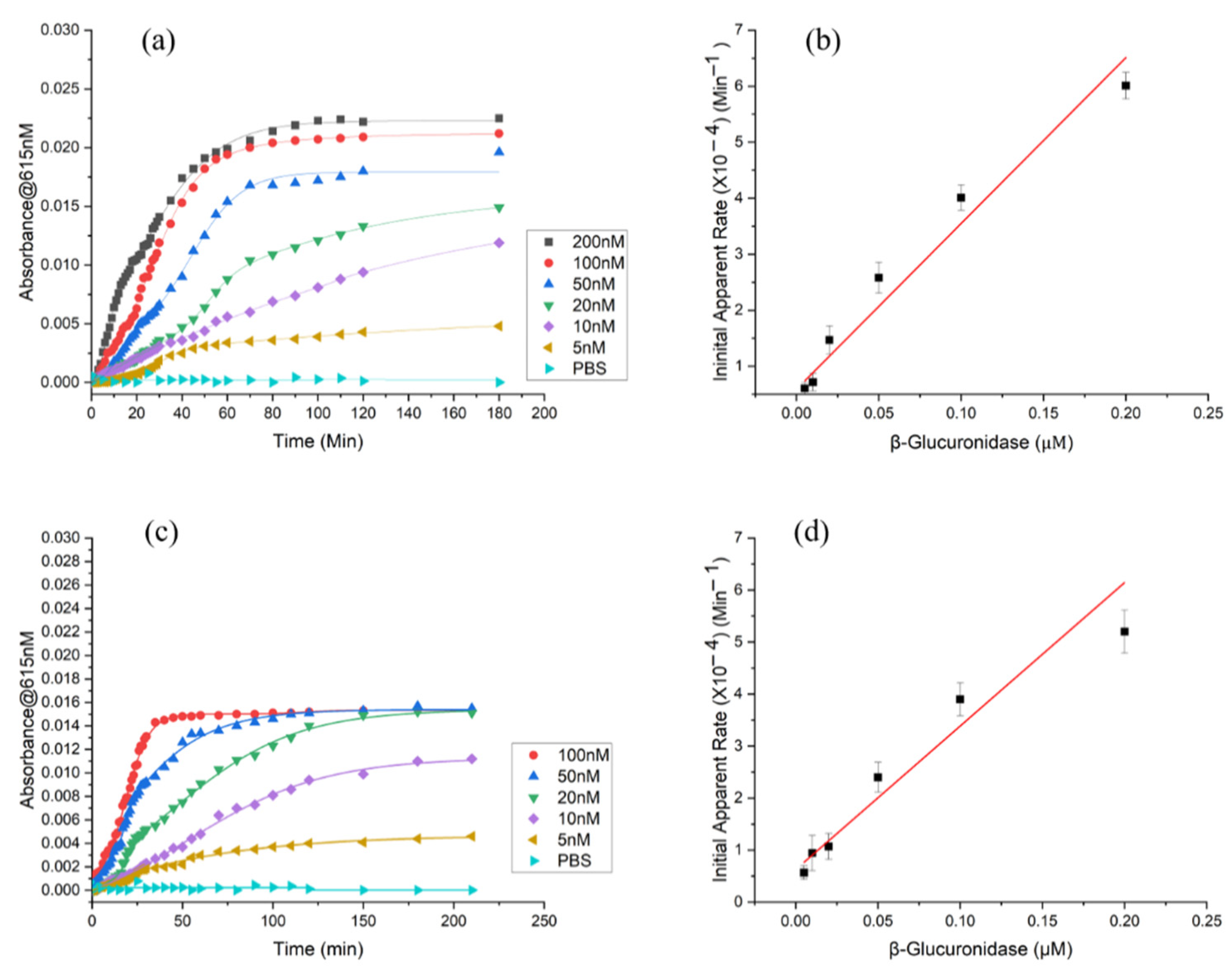
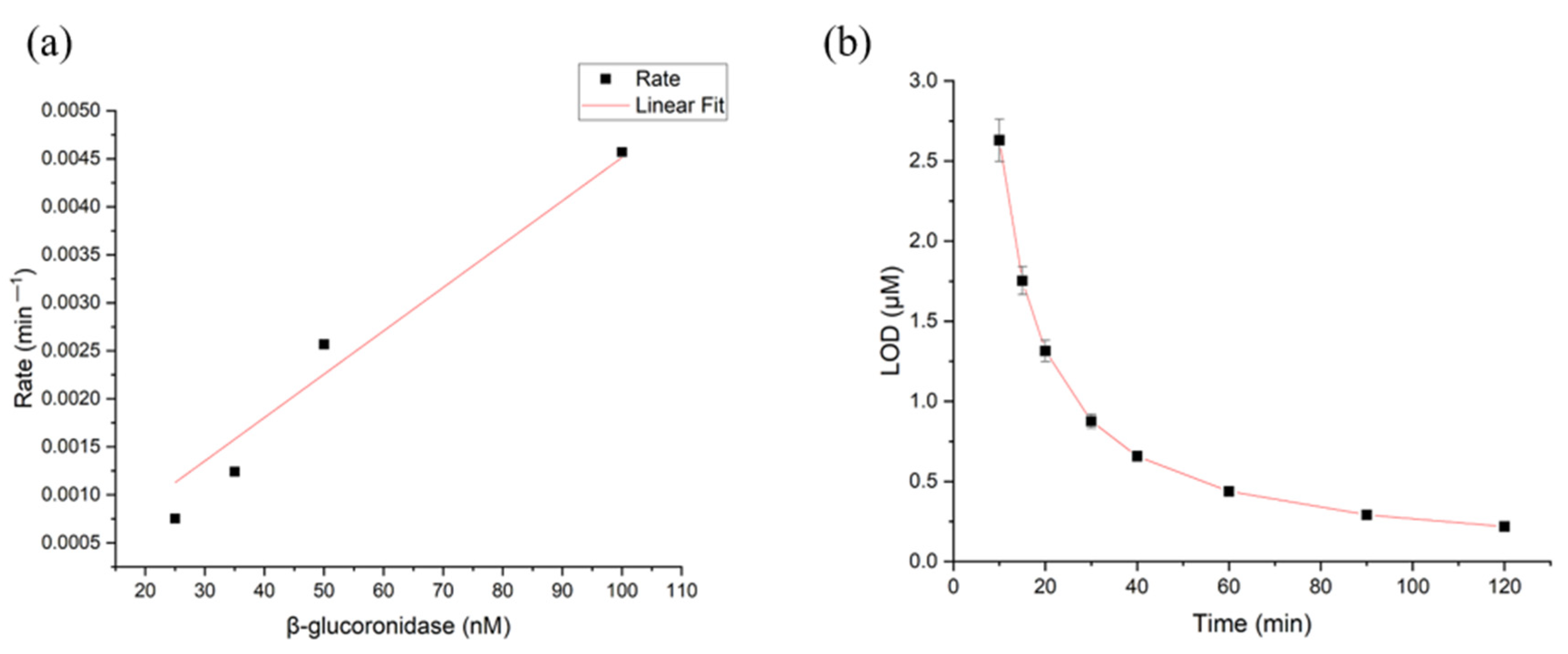
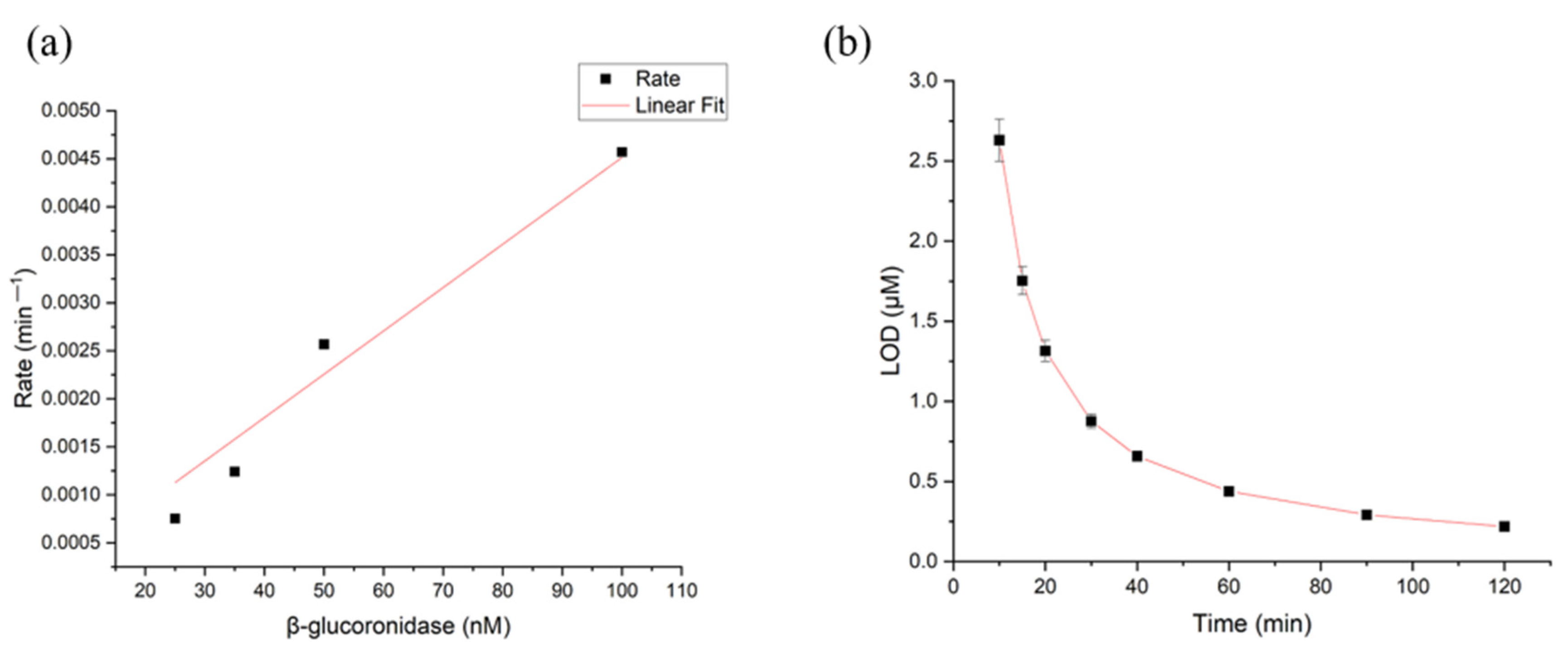
3.4. Limit of Detection (LOD) and Limit of Quantification (LOQ) for β-Glucuronidase in CS/PEO NFs and PUAA-ADH MS Hydrogel Films
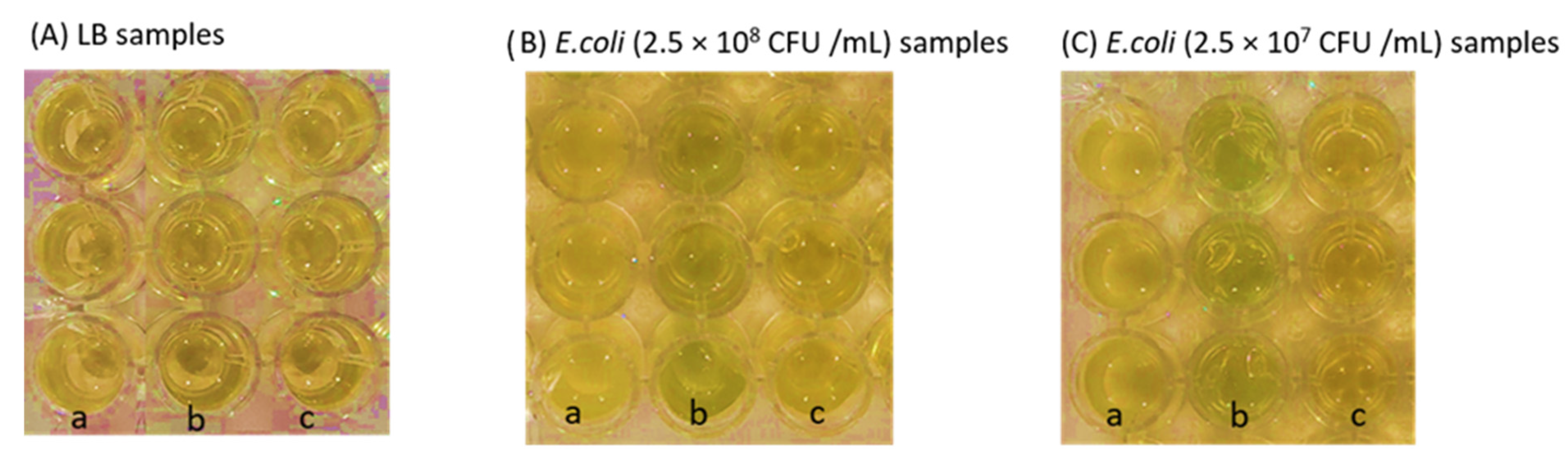
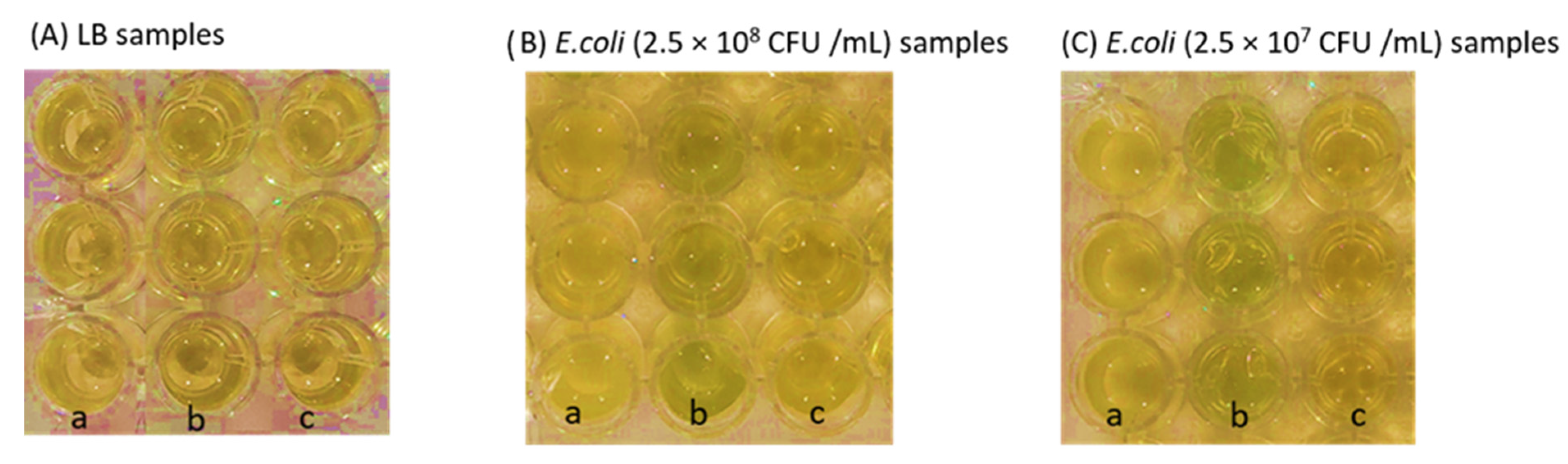
3.5. Detection Performance of PUAA-ADH_X-Gluc Films and CS/PEO Nanofibers with Bacterial Cultures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Aliabadi, S.; Anyanwu, P.; Beech, E.; Jauneikaite, E.; Wilson, P.; Hope, R.; Majeed, A.; Muller-Pebody, B.; Costelloe, C. Effect of Antibiotic Stewardship Interventions in Primary Care on Antimicrobial Resistance of Escherichia coli bacteraemia in England (2013–18): A Quasi-experimental, Ecological, Data Linkage Study. Lancet Infect. Dis. 2021, 21, 1689–1700. [Google Scholar] [CrossRef]
- Firoozeh, F.; Zibaei, M.; Badmasti, F.; Khaledi, A. Virulence Factors, Antimicrobial Resistance and the Relationship Between these Characteristics in Uropathogenic Escherichia coli. Gene Rep. 2022, 27, 101622. [Google Scholar] [CrossRef]
- Shaibani, P.M.; Etayash, H.; Jiang, K.; Sohrabi, A.; Hassanpourfard, M.; Naicker, S.; Sadrzadeh, M.; Thundat, T. Portable Nanofiber-light Addressable Potentiometric Sensor for Rapid Escherichia coli Detection in Orange Juice. ACS Sens. 2018, 3, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Hur, H.-G.; Sadowsky, M.J.; Byappanahalli, M.N.; Yan, T.; Ishii, S. Environmental Escherichia coli: Ecology and public health implications—A review. J. Appl. Microbiol. 2017, 123, 570–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orenga, S.; James, A.L.; Manafi, M.; Perry, J.D.; Pincus, D.H. Enzymatic Substrates in Microbiology. J. Microbiol. Methods 2009, 79, 139–155. [Google Scholar] [CrossRef]
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-Guided Discovery of Antibiotics that Inhibit Peptidoglycan Remodelling. Nature 2020, 578, 582–587. [Google Scholar] [CrossRef]
- Gall, T.L.; Lemercier, G.; Chevreux, S.; Tücking, K.S.; Ravel, J.; Thetiot, F.; Jonas, U.; Schönherr, H.; Montier, T. Ruthenium(II) Polypyridyl Complexes as Photosensitizers for Antibacterial Photodynamic Therapy: A Structure-Activity Study on Clinical Bacterial Strains. Chem. Med. Chem. 2018, 13, 2229–2239. [Google Scholar] [CrossRef]
- Zhao, X.; Lin, C.W.; Wang, J.; Oh, D.H. Advances in Rapid Detection Methods for Foodborne Pathogens. J. Microbiol. Biotechnol. 2014, 24, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Deusenbery, C.; Wang, Y.; Shukla, A. Recent Innovations in Bacterial Infection Detection and Treatment. ACS Infect. Dis. 2021, 7, 695–720. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, P.; Nienhuser, A.; Packroff, G.; Wernicke, F.; Mehling, A.; Nixdorf, K.; Fiedler, S.; Kolauch, C.; Esser, M. Molecular Identification of Coliform Bacteria Isolated from Drinking Water Reservoirs with Traditional Methods and the Colilert-18 System. Int. J. Hyg. Environ. Health 2007, 211, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Haller, L.; Hutton, G.; Bartram, J. Estimating the Costs and Health Benefits of Water and Sanitation Improvements at Global Level. J. Water Health 2007, 5, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Mukherjee, M.D.; Sumana, G.; Gupta, R.K.; Sood, S.; Malhotra, B.D. Biosensors for Pathogen Detection: A Smart Approach Towards Clinical Diagnosis. Sens. Actuators B 2014, 197, 385–404. [Google Scholar] [CrossRef]
- Cui, C.; Shu, W.; Li, P. Fluorescence In Situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016, 4, 89. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhou, Y.; Fu, J.; Fang, H.; Li, Y.; Huang, X.; Xiong, Y. A Self-luminous Bifunctional Bacteria Directed Fluorescent Immunosensor for the Simultaneous Detection and Quantification of Three Pathogens in Milk. Sens. Actuators B Chem. 2021, 338, 129757–129764. [Google Scholar] [CrossRef]
- Sylvia, S.V.; Salomi, R.J.; Rajendran, L.; Lyons, M.E.G. Amperometric Biosensors and Coupled Enzyme Nonlinear Reactions Processes: A Complete Theoretical and Numerical Approach. Electrochim. Acta 2022, 415, 140236. [Google Scholar] [CrossRef]
- Radke, S.M.; Alocilja, E.C. A High Density Microelectrode Array Biosensor for Detection of E. coli O157:H7. Biosens. Bioelectron. 2005, 20, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, S.; Lee, H.; Lee, S.; Kim, S.; Lee, J.; Ha, J.; Oh, H.; Lee, Y.; Kim, Y.; et al. Rapid Detection of Escherichia coli in Fresh Foods Using a Combination of Enrichment and PCR Analysis. Korean J. Food Sci. Anim. Resour. 2018, 38, 829–834. [Google Scholar]
- Zhou, J.; Loftus, A.L.; Mulley, G.; Jenkins, A.T.A. A Thin Film Detection/Response System for Pathogenic Bacteria. J. Am. Chem. Soc. 2010, 132, 6566–6570. [Google Scholar] [CrossRef]
- Marshall, S.E.; Hong, S.-H.; Thet, N.T.; Jenkins, A.T.A. Effect of Lipid and Fatty Acid Composition of Phospholipid Vesicles on Long-Term Stability and Their Response to Staphylococcus Aureus and Pseudomonas aeruginosa Supernatants. Langmuir 2013, 29, 6989–6995. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Hain, N.; Raoufi, M.; Handschuh-Wang, S.; Wang, T.; Jiang, X.; Schönherr, H. Enzyme Degradable Polymersomes from Hyaluronic Acid-block-Poly(ε-Caprolactone) Copolymers for the Detection of Enzymes of Pathogenic Bacteria. Biomacromolecules 2015, 16, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Tücking, K.S.; Grützner, V.; Unger, R.E.; Schönherr, H. Dual Enzyme-Responsive Capsules of Hyaluronic Acid-block-Poly(lactic acid) for Sensing Bacterial Enzymes. Macromol. Rapid Comm. 2015, 36, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Miranda, O.R.; Li, X.; Garcia-Gonzalez, L.; Zhu, Z.J.; Yan, B.; Bunz, U.H.F.; Rotello, V.M. Colorimetric Bacteria Sensing Using a Supramolecular Enzyme-Nanoparticle Biosensor. J. Am. Chem. Soc. 2011, 133, 9650–9653. [Google Scholar] [CrossRef] [Green Version]
- Horne, J.; McLoughlin, L.; Bridgers, B.; Wujcik, E.K. Recent Developments in Nanofiber-Based Sensors for Disease Detection, Immunosensing, and Monitoring. Sens. Act. Rep. 2020, 2, 100005–100013. [Google Scholar] [CrossRef]
- Aliheidari, N.; Aliahmad, N.; Agarwal, M.; Dalir, H. Electrospun Nanofibers for Label-Free Sensor Applications. Sensors 2019, 19, 3587. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, M.M.S.; Laabei, M.; Jenkins, A.T.A.; Schönherr, H. Autonomously Sensing Hydrogels for the Rapid and Selective Detection of Pathogenic Bacteria. Macromol. Rapid Commun. 2015, 36, 2123–2128. [Google Scholar] [CrossRef]
- Jia, Z.; Sukker, I.; Müller, M.; Schönherr, H. Selective Determination of Key Enzymes of Pathogenic and Nonpathogenic Bacteria on Autonomously Reporting Shape-Encoded Hydrogel Patterns. ACS Appl. Mater. Interfaces 2018, 10, 5175–5184. [Google Scholar] [CrossRef]
- Kaur, K.; Chelangat, W.; Druzhinin, S.I.; Karuri, N.W.; Müller, M.; Schönherr, H. Quantitative E. coli Enzyme Detection in Reporter Hydrogel-Coated Paper Using a Smartphone Camera. Biosensors 2021, 11, 25. [Google Scholar] [CrossRef]
- Ebrahimi, M.M.S.; Schönherr, H. Enzyme-Sensing Chitosan Hydrogels. Langmuir 2014, 30, 7842–7850. [Google Scholar] [CrossRef]
- Das, D.; Alhusaini, Q.F.M.; Kaur, K.; Raoufi, M.; Schönherr, H. Enzyme-Responsive Biopolymeric Nanogel Fibers by Extrusion: Engineering of High-Surface-Area Hydrogels and Application in Bacterial Enzyme Detection. ACS Appl. Mater. Interfaces 2021, 13, 12928–12940. [Google Scholar] [CrossRef]
- Halicka, K.; Cabaj, J. Electrospun Nanofibers for Sensing and Biosensing Applications—A Review. Int. J. Mol. Sci. 2021, 22, 6357. [Google Scholar] [CrossRef]
- Kang, S.; Zhao, K.; Yu, D.G.; Zheng, X.; Huang, C. Advances in Biosensing and Environmental Monitoring Based on Electrospun Nanofibers. Adv. Fiber Mater. 2022, 4, 404–435. [Google Scholar] [CrossRef]
- Liao, Y.; Loh, C.H.; Tian, M.; Wang, R.; Fane, A.G. Progress in Electrospun Polymeric Nanofibrous Membranes for Water Treatment: Fabrication, Modification and Applications. Prog. Polym. Sci. 2018, 77, 69–94. [Google Scholar] [CrossRef]
- Zhou, K.; Wang, M.; Zhou, Y.; Sun, M.; Xie, Y.; Yu, D.G. Comparisons of Antibacterial Performances Between Electrospun Polymer@Drug Nanohybrids with Drug-Polymer Nanocomposites. Adv. Compos. Hybrid. Mater. 2022, 5, 907–919. [Google Scholar] [CrossRef]
- Wen, P.; Wen, Y.; Zong, M.H.; Linhardt, R.J.; Wu, H. Encapsulation of Bioactive Compound in Electrospun Fibers and Its Potential Application. J. Agric. Food Chem. 2017, 65, 9161–9179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Jia, J.; Wang, J. Nonenzymatic Glucose Sensor Based on Graphene Oxide and Electrospun NiO Nanofibers. Sens. Actuators B 2012, 171, 580–587. [Google Scholar] [CrossRef]
- Migliorini, F.L.; Sanfelice, R.C.; Mercante, L.A.; Andre, R.S.; Mattoso, L.H.C.; Correa, D.S. Urea Impedimetric Biosensing Using Electrospun Nanofibers Modified with Zinc Oxide Nanoparticles. Appl. Surf. Sci. 2018, 443, 18–23. [Google Scholar] [CrossRef]
- Tripathy, S.; Bhandari, V.; Sharma, P.; Vanjari, S.R.K.; Singh, S.G. Chemiresistive DNA Hybridization Sensor with Electrospun Nanofibers: A Method to Minimize Inter-Device Variability. Biosens. Bioelectron. 2019, 133, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, L.; Liu, J.; Li, C. A Flexible Bimodal Sensor Based on an Electrospun Nanofibrous Structure for Simultaneous Pressure-Temperature Detection. Nanoscale 2019, 11, 14242–14249. [Google Scholar] [CrossRef] [PubMed]
- Thiha, A.; Ibrahim, F.; Muniandy, S.; Dinshaw, I.J.; The, S.J.; Thong, K.L.; Leo, B.F.; Madou, M. All-Carbon Suspended Nanowire Sensors as a Rapid Highly-Sensitive Label-Free Chemiresistive Biosensing Platform. Biosens. Bioelectron. 2018, 107, 145–152. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.; Saldana, L.; Ackerman, D.N.; Su, Y.; John, J.V.; Chen, S.; Weihs, S.; Reid, S.P.; Satrapi, J.-L.; Carlson, M.A.; et al. Ultra-Absorptive Nanofiber Swabs for Improved Collection and Test Sensitivity of SARS-CoV-2 and other Biological Specimens. Nano Lett. 2021, 21, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Adabi, M.; Esnaashari, S.S.; Adabi, M. An Electrochemical Immunosensor Based on Electrospun Carbon Nanofiber Mat Decorated with Gold Nanoparticles and Carbon Nanotubes for the Detection of Breast Cancer. J. Porous Mater. 2021, 28, 415–421. [Google Scholar] [CrossRef]
- Truskewycz, A.; Truong, V.K.; Ball, A.S.; Houshyar, S.; Nassar, N.; Yin, H.; Murdoch, B.J.; Cole, I. Fluorescent Magnesium Hydroxide Nanosheet Bandages with Tailored Properties for Biocompatible Antimicrobial Wound Dressings and pH Monitoring. ACS Appl. Mater. Inter. 2021, 13, 27904–27919. [Google Scholar] [CrossRef] [PubMed]
- Chagas, P.A.M.; Schneider, R.; Santos, D.M.; Otuka, A.J.G.; Mendonca, C.R.; Correa, D.S. Bilayered Electrospun Members Composed of Poly(lactic-acid)/Natural Rubber: A Strategy Against Curcumin Photodegradation for Wound Dressing Application. React. Funct. Polym. 2021, 163, 104889–104899. [Google Scholar] [CrossRef]
- Atashgah, R.; Ghasemi, A.; Raoufi, M.; Abdollahifar, M.; Zanganeh, S.; Nejadnik, H.; Abdollahi, A.; Sharifi, S.; Lea, B.; Cuerva, M.; et al. Restoring Endogenous Repair Mechanisms to Heal Chronic Wounds with a Multi-Functional Wound Dressing. Mol. Pharm. 2021, 18, 3171–3180. [Google Scholar] [CrossRef]
- Song, Q.; Steuber, M.; Druzhinin, S.I.; Schönherr, H. Tailored Combinatorial Microcompartments through the Self-Organization of Microobjects: Assembly, Characterization, and Cell Studies. Angew. Chem. 2019, 131, 5300–5304. [Google Scholar] [CrossRef]
- Hillborg, H.; Tomczak, N.; Olàh, A.; Schönherr, H.; Vancso, J.G. Nanoscale Hydrophobic Recovery: A Chemical Force Microscopy Study of UV/Ozone—Treated Crosslinked Poly(dimethylsiloxane). Langmuir 2004, 20, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.M.; Bossard, F.; Rinaudo, M. Preparation of Pure and Stable Chitosan Nanofibers by Electrospinning in the Presence of Poly(ethylene oxide). Int. J. Mol. Sci. 2016, 17, 1790–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goonoo, N.; Khanbabaee, B.; Steuber, M.; Bhaw-Luximon, A.; Jonas, U.; Pietsch, U.; Jhurry, D.; Schonherr, H. κ-Carrageenan Enhances the Biomineralization and Osteogenic Differentiation of Electrospun Polyhydroxybutyrate and Polyhydroxybutyrate Valerate Fibers. Biomacromolecules 2017, 18, 1563–1573. [Google Scholar] [CrossRef]
- Chen, S.; Xu, X.L.; Zhou, B.; Tian, J.; Luo, B.M.; Zhang, L.M. Acidic pH-Activated Gas-Generating Nanoparticles with Pullulan Decorating for Hepatoma-Targeted Ultrasound Imaging. ACS Appl. Mater. Interfaces 2019, 11, 22194–22205. [Google Scholar] [CrossRef]
- Rohindra, D.R.; Nand, A.V.; Khurma, J.R. Swelling Properties of Chitosan Hydrogels. South Pacific J. Nat. Appl. Sci. 2004, 22, 32–35. [Google Scholar] [CrossRef]
- Jin, J.; Song, M. Chitosan and Chitosan-PEO Blend Membranes Crosslinked by Genipin for Drug Release. J. Appl. Polym. Sci. 2006, 102, 436–444. [Google Scholar] [CrossRef]
- Mahardian, A. Biocompatible Hydrogel Film of Polyethylene Oxide-Polyethylene Glycol Dimetacrylate for Wound Dressing Application. IOP Conf. Ser.: Mater. Sci. Eng. 2018, 288, 012076. [Google Scholar]
- Mirzaei, E.; Sarkar, S.; Rezayat, S.M.; Faridi-Majidi, R. Herbal Extract Loaded Chitosan-Based Nanofibers as a Potential Wound-Dressing. J. Adv. Med. Sci. Appl. Technol. 2016, 2, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.J.; Cates, R.S.; Stroot, P.G.; Toomey, R. Swelling-Induced Instabilities in Microscale, Surface-Confined Poly(N-isopropylacryamide) Hydrogels. Soft Matter 2010, 6, 3876–3882. [Google Scholar] [CrossRef]
- Pakravan, M.; Heuzey, M.C.; Ajji, A. A Fundamental Study of Chitosan/PEO Electrospinning. Polymer 2011, 52, 4813–4824. [Google Scholar] [CrossRef]
- Otero, T.F.; Martinez, J.G. Structural and Biomimetic Chemical Kinetics: Kinetic Magnitudes Include Structural Information. Adv. Funct. Mater. 2013, 23, 404–416. [Google Scholar] [CrossRef]
- Tzafriri, A.R. Michaelis-Menten Kinetics at High Enzyme Concentrations. Bull. Math. Biol. 2003, 65, 1111–1129. [Google Scholar] [CrossRef]
- Ebrahimi, M.M.S.; Voss, Y.; Schönherr, H. Rapid Detection of Escherichia coli via Enzymatically Triggered Reactions in Self-Reporting Chitosan Hydrogels. ACS Appl. Mater. Interfaces 2015, 7, 20190–20199. [Google Scholar]
- Kasaai, M.R. A Review of Several Reported Procedures to Determine the Degree of N-Acetylation for Chitin and Chitosan Using Infrared Spectroscopy. Carbohydr. Polym. 2008, 71, 497–508. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinyua, C.K.; Owino, A.O.; Kaur, K.; Das, D.; Karuri, N.W.; Müller, M.; Schönherr, H. Impact of Surface Area on Sensitivity in Autonomously Reporting Sensing Hydrogel Nanomaterials for the Detection of Bacterial Enzymes. Chemosensors 2022, 10, 299. https://doi.org/10.3390/chemosensors10080299
Kinyua CK, Owino AO, Kaur K, Das D, Karuri NW, Müller M, Schönherr H. Impact of Surface Area on Sensitivity in Autonomously Reporting Sensing Hydrogel Nanomaterials for the Detection of Bacterial Enzymes. Chemosensors. 2022; 10(8):299. https://doi.org/10.3390/chemosensors10080299
Chicago/Turabian StyleKinyua, Celestine Kathure, Ayub Omondi Owino, Kawaljit Kaur, Dipankar Das, Nancy Wangechi Karuri, Mareike Müller, and Holger Schönherr. 2022. "Impact of Surface Area on Sensitivity in Autonomously Reporting Sensing Hydrogel Nanomaterials for the Detection of Bacterial Enzymes" Chemosensors 10, no. 8: 299. https://doi.org/10.3390/chemosensors10080299
APA StyleKinyua, C. K., Owino, A. O., Kaur, K., Das, D., Karuri, N. W., Müller, M., & Schönherr, H. (2022). Impact of Surface Area on Sensitivity in Autonomously Reporting Sensing Hydrogel Nanomaterials for the Detection of Bacterial Enzymes. Chemosensors, 10(8), 299. https://doi.org/10.3390/chemosensors10080299